오늘은 NGS 분석과 관련하여 인코렌탈이라는 생물정보 분석 솔루션 단기 임대 서비스에 대해 알아보겠습니다. 평소에 NGS 데이터를 통해 연구하거나 분석을 진행하는 분들께 유용한 서비스일 것 같아 소개하게 되었습니다.
1. 인코렌탈이란?
㈜인실리코젠에서는 연구자들이 생물정보를 더욱 편리하게 분석할 수 있도록 도와주는 생물정보 분석 솔루션(IPA with Analysis Match, CLC Genomics Workbench 등)을 공급하고 있습니다.
인코렌탈은 생물정보 분석 솔루션을 원하는 만큼 유연하게 대여할 수 있는 서비스로 직접 코딩하지 못하더라도 GUI 환경에서 간편하게 사용할 수 있다는 게 정말 큰 장점입니다.
2. 인코렌탈을 이용한 RNA seq 분석 및 결과 Review
인코렌탈을 이용하여 분석할 데이터는 SRA에 등재된 RNA Seq 데이터입니다. Ovarian cancer tumor 데이터로 Fastq 데이터로 다운로드 받아서 이번 분석에 사용했습니다.
SRA Archive: NCBI
Updated: Wed Dec 6 10:54:10 EST 2023
trace.ncbi.nlm.nih.gov

오늘 진행할 분석은 RNA 분석으로 해당 데이터에 어떤 유전자들이 발현되고 있는지 확인해볼 예정입니다.
RNA seq 분석은 생각보다 데이터가 작아서 시간이 적게 소요되는데 인코렌탈은 단기 사용도 가능하므로 필요한 분석에 효율적으로 이용할 수 있다는 점이 좋은 것 같다고 느꼈습니다.
3. RNA seq 분석 진행 방법
<분석 데이터 준비하기>
먼저 분석하기 위해 위와 같이 Fastq 같은 raw 데이터를 업로드 해야 합니다.
파일 업로드를 위해서는 FileZilla라는 프로그램을 먼저 설치해야 합니다.
1) Remote Ripple 다운로드
2) CLC Genomics Workbench 실행
● CLC Genomics Workbench란?
: QIAGEN에서 만든 Next Generation Sequencing (NGS) 데이터 분석을 위한 GUI 기반의 소프트웨어 입니다. Sanger/ Illumina/ Ion Torrent/ PacBio/ Nanopore 등 다양한 NGS 포맷을 지원하고 있다는 장점이 있으며 SIMD 기술을 적용한 초고속 NGS 데이터 분석이 가능합니다.

위와 같이 다양한 연구에 통합적으로 사용할 수 있는 Tool을 제공하며, Genomics/ Transcriptomics/ Epigenomics/ Metagenomics 등 다양한 분야의 응용이 가능합니다.

3) FileZilla 설치 및 실행 (데이터 업로드)

File Zilla를 설치해주고 아래와 같이 부여받은 ID, PW로 서버에 로그인해서 사용할 데이터를 업로드 시켜줍니다.
File Ziilla를 설치하면 프로토콜 및 호스트 정보를 입력하라고 하는 창이 뜨는데, 이때 인실리코젠에서 부여받은 계정을 입력해주면 됩니다.

위와 같이 다시 접속되면 연결된 것입니다. 그리고 새 사이트에 설정해준 이름을 클릭합니다.
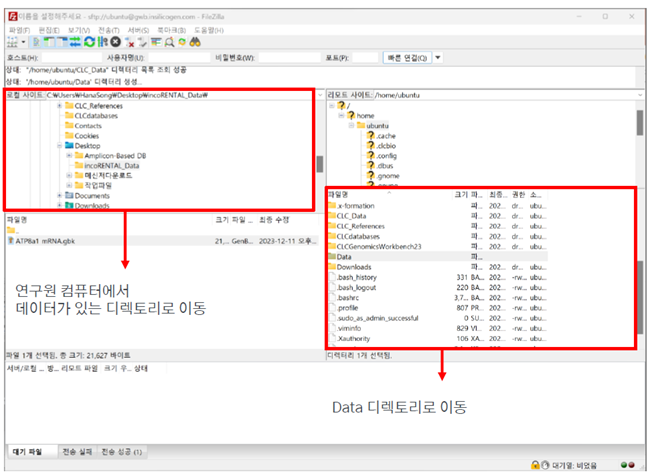
필요한 데이터를 개인 컴퓨터에서(왼쪽 화면) /home/ubuntu 로 드래그하여 옮겨줍니다.(오른쪽)
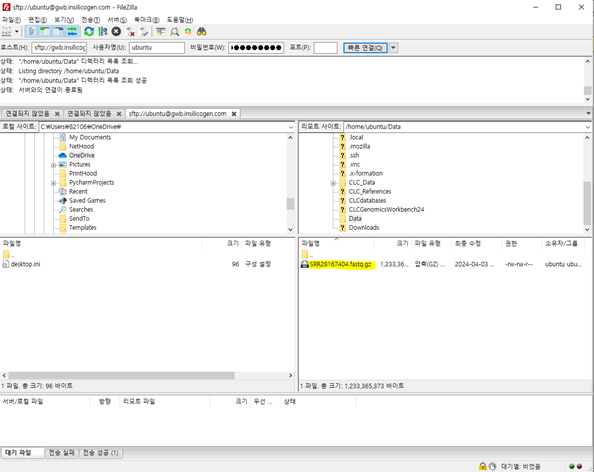
이렇게 분석에 사용될 data를 서버에 업로드 해주었습니다.
4) CLC Genomics Workbench 데이터 가져오기
● 분석용 data 업로드
①일반적인 data 업로드 방법
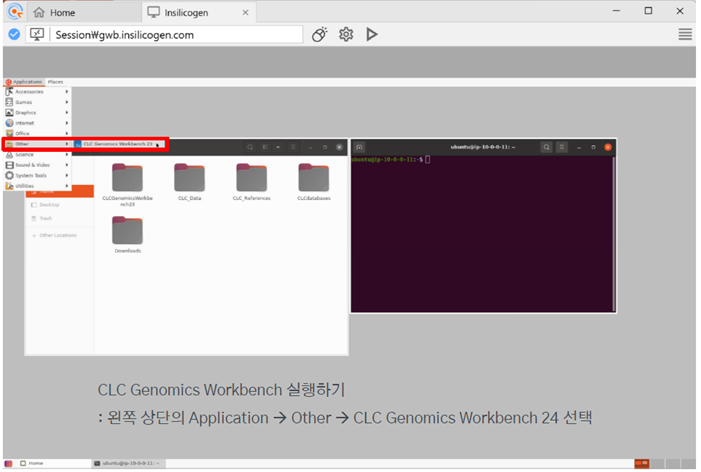
CLC Genomics Workbench 24를 선택해서 실행시켜줍니다.
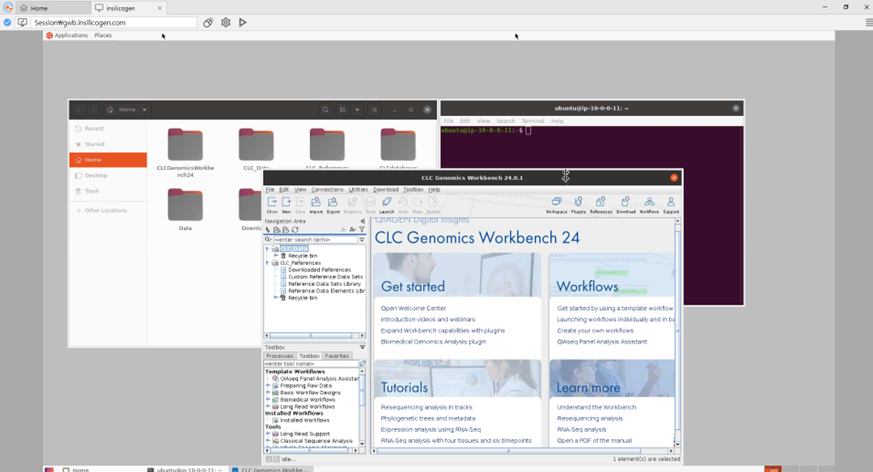
접속하면 다음과 같은 창이 뜹니다.
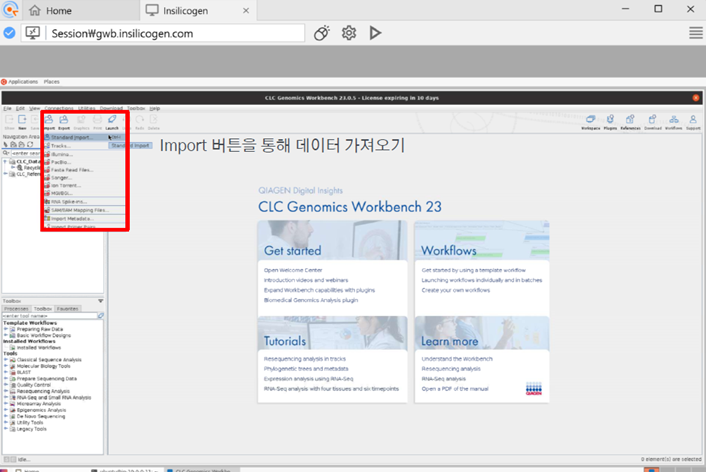
Import 버튼을 통하여 데이터를 가져옵니다.
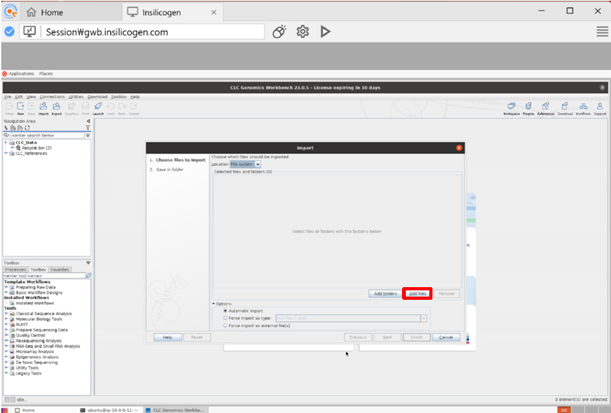
Add files를 선택하고
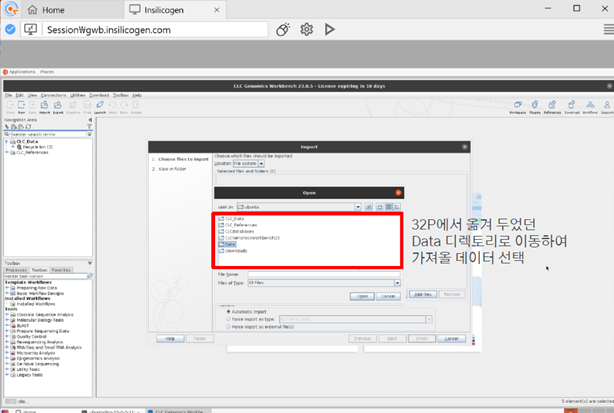
FileZilla를 통해 옮겨두었던 데이터를 선택해서 import 해줍니다.
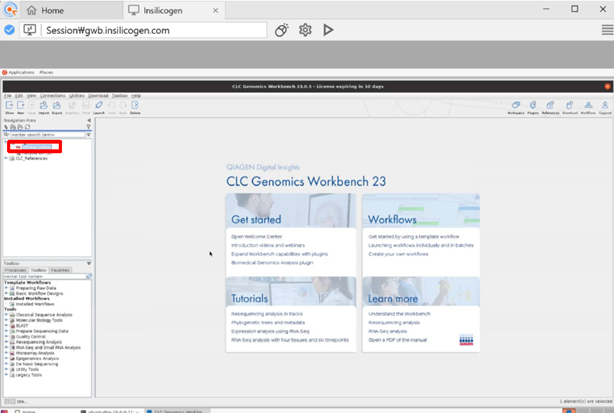
Import가 완료되면 왼쪽 상단 박스에 업로드 해준 샘플이 생긴 것을 확인할 수 있습니다.
② Fastq 업로드 방법
Fastq를 업로드 하고 싶다면 아래와 같은 방법으로 업로드 해주면 됩니다.
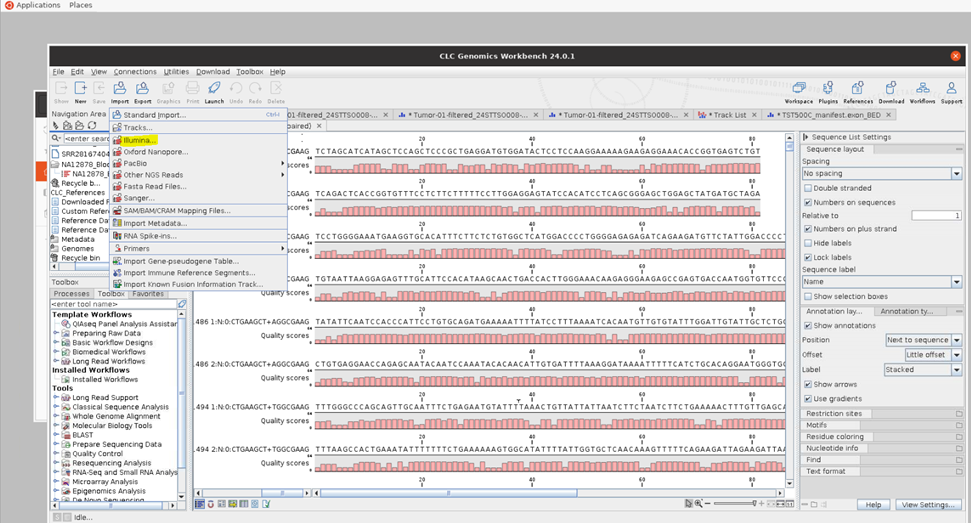
Import 탭에서 Fastq를 분석한 시퀀서 종류를 골라줍니다. 일루미나 장비를 사용하였으므로 illumina를 선택하였습니다.
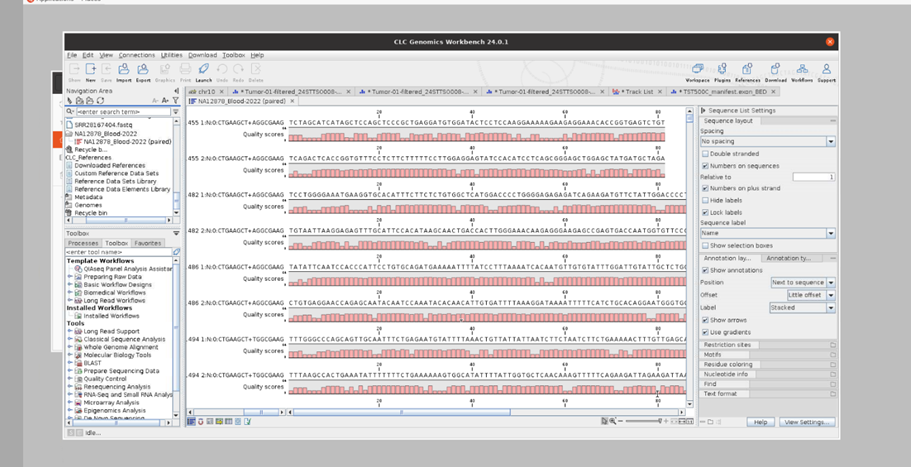
Fastq에서는 Quality score, 서열에 대한 정보를 위와 같은 정보들을 확인할 수 있습니다.
③ VCF 업로드 방법
VCF 업로드를 통하여 Variant calling 결과도 확인할 수 있습니다.
5) Reference Download
그다음 분석에 필요한 부가적인 파일들을 다운로드 받아보겠습니다.
① reference 받기
사람 RNA seq 데이터이므로 human hg19로 reference genome을 받아줍니다. CLC Genomics Workbench에서 우측 상단에 References라고 되어있는 부분을 누르면 연구자들이 주로 사용하는 human, mouse 등 데이터를 쉽게 다운로드 받을 수 있습니다.
저는 human 데이터이므로 hg19 reference를 받았습니다.
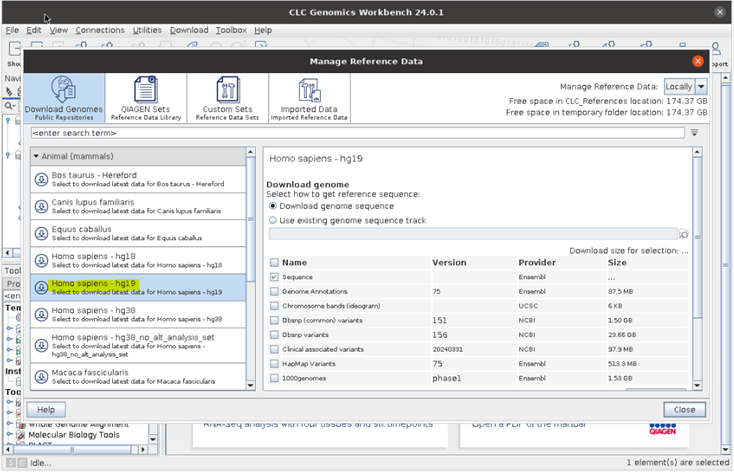
Homo sapiens-hg19를 선택해서 human hg19 reference를 받았습니다.
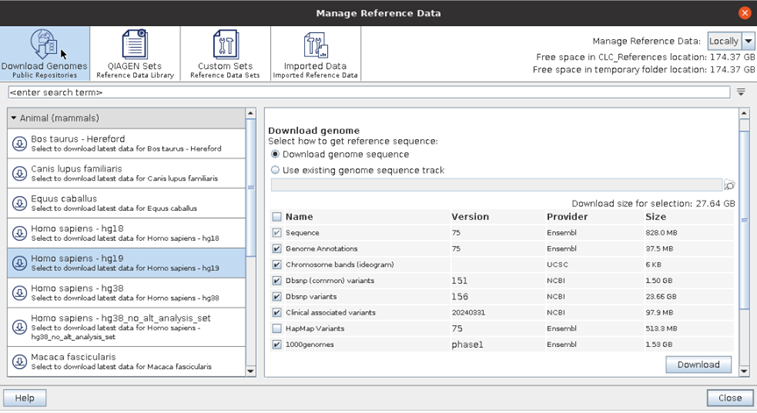
NCBI, UCSC 등 다양한 DB를 다운로드 받을 수 있습니다. 선택했다면 Download 버튼을 누릅니다.
이 작업은 시간이 오래 소요되니 꼭 필요한 DB만 다운로드 받으면 좋을 것 같습니다. 데이터가 이미 서버상에 올라와 있어 reference를 web 페이지에서 따로 찾을 필요가 없어서 좋았습니다.
② RNA-seq analysis with CLC Genomic Workbench 분석
RNA-seq의 전반적인 분석 과정은 [Data import/ Reference] – [Preprocessing] – [RNA-seq analysis] – [Visualization] 입니다.
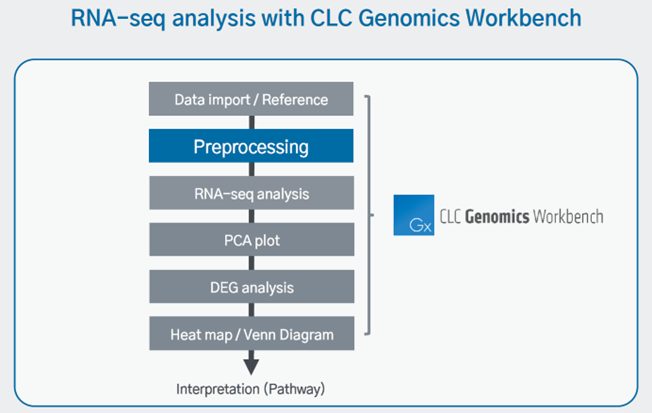
위에서 필요한 Data 들은 import 해왔기 때문에 preprocessing부터 시작합니다.
6) Preprocessing
Preprocessing 단계는 trimming 하는 단계로 NGS 분석 시, contamination 되거나 낮은 퀄리티의 데이터를 제거하는 작업을 의미합니다.
즉, 시퀀싱 한 raw 데이터는 trimming 단계를 거쳐서 clean 한 데이터로 생성됩니다.
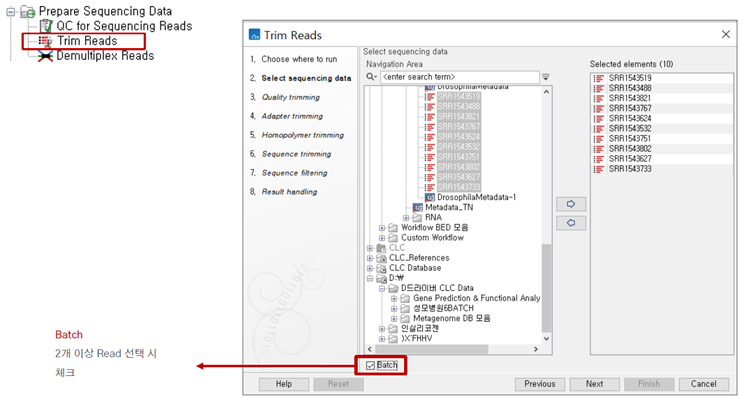
Trimming할 데이터를 골라줍니다.
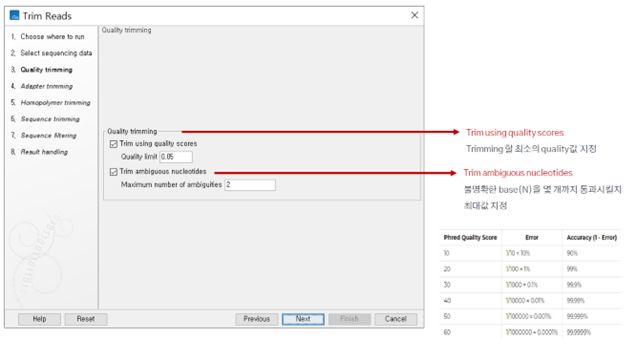
이후 Quality trimming 시 지표로 사용할 score를 정해줍니다.
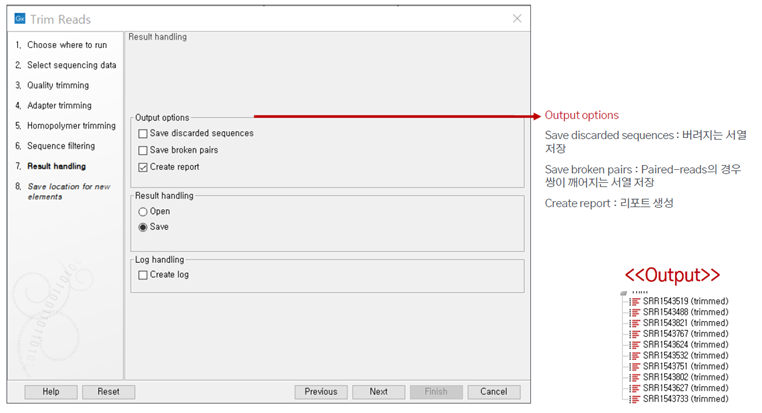
그리고 최종 데이터를 어떻게 저장할지 지정해주면 trimming 분석이 진행됩니다.
7) Trimming Result

Trimming 결과는 Trim summary, Read length, Trim setting, Detailed trim result, Automatic adapter read-through trimming에 대한 정보를 확인할 수 있습니다.
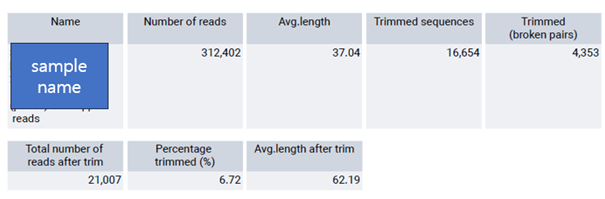
이번에 사용한 데이터의 경우, Read 수가 312,402개였는데 trimming이 된 sequence 들이 16654였던 것을 볼 수 있습니다.
trimming이후에 total read 수는 21,007개로 감소한 것을 확인할 수 있었습니다.

이렇게 Trimming 시 세팅했었던 limit 기준도 확인할 수 있습니다. 저는 low quality sequence limit을 기본 값이었던 0.05로 설정하였습니다.

자세한 trim result도 확인할 수 있는데요, quality로 인해서 trimming 된 read의 개수도 알 수 있으며 이번에 5,466개가 quality fail로 인해서 trimming 되었습니다.
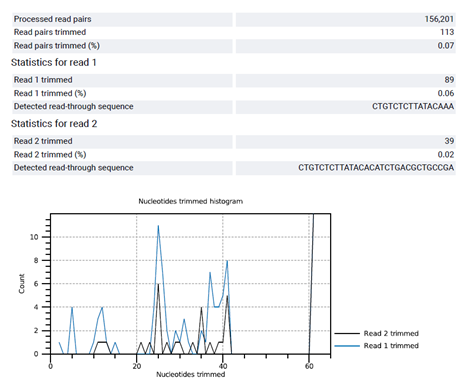
이와 같이 trimming이 얼마나 되었고, trimming 된 이전과 이후를 비교해볼 수 있습니다.
8) RNA-seq analysis
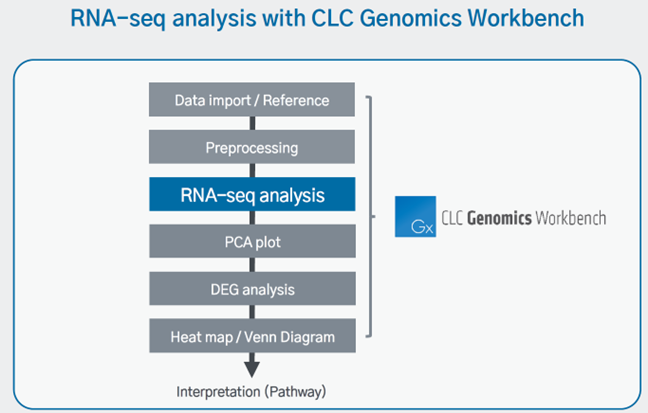
Trimming 완료 후 trimming 된 데이터를 이용하여 RNA-seq 분석을 진행합니다.
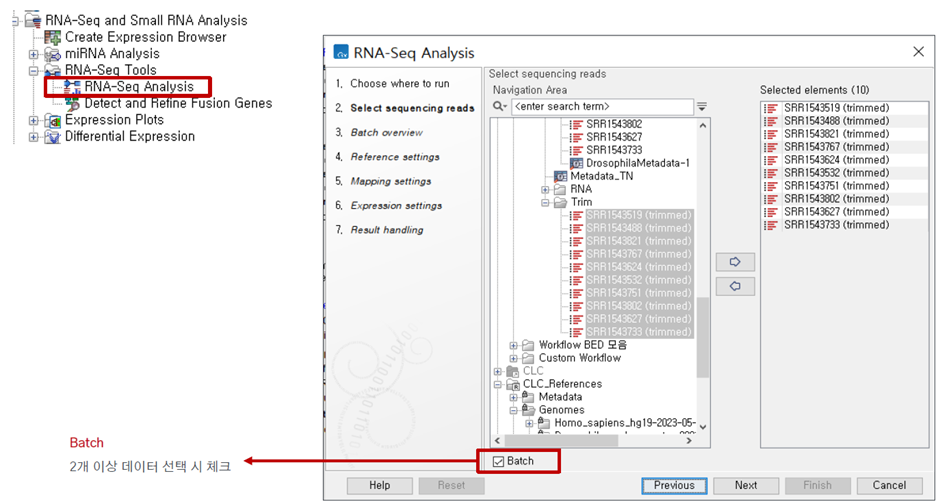
분석에 사용할 Trimming 된 데이터를 선택해줍니다.
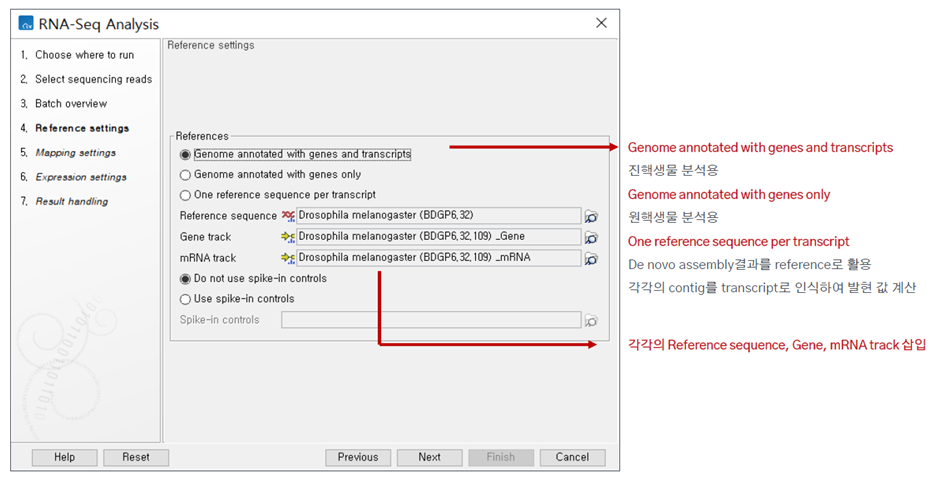
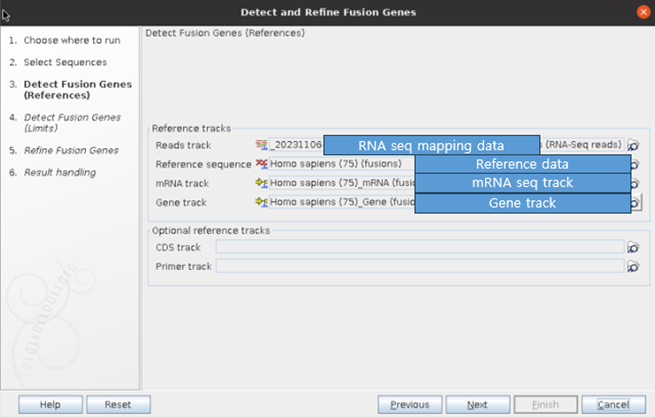
사용하는 데이터의 reference 데이터, Gene track, mRNA track 데이터를 함께 지정해줍니다. 저는 reference 데이터로 hg19를 사용하였습니다. Gene track, mRNA track 데이터는 reference 다운로드 시 함께 다운로드 되니 따로 준비할 필요는 없었습니다.
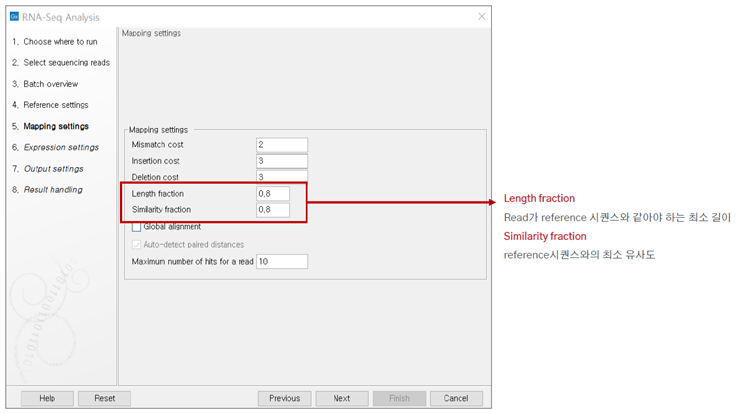
Read 가 reference 시퀀스와 같아야 하는 최소길이를 지정해주고 similarity fraction도 설정해줍니다. CLC Genomics Workbench에서 권장하고 있는 기본값으로 분석 진행하였습니다.
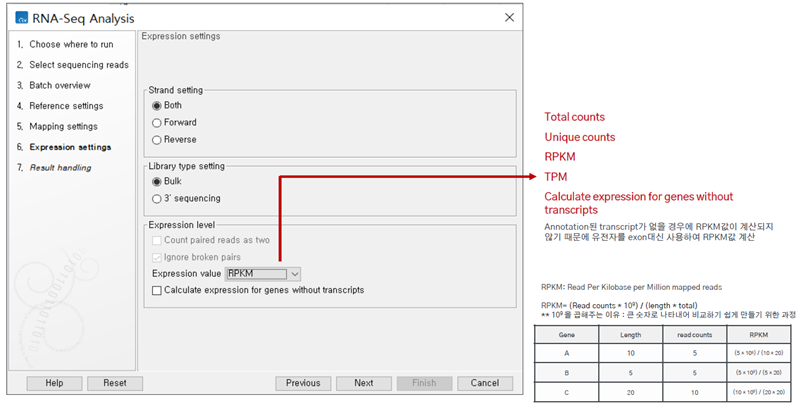
마지막으로 Strand setting을 해주며 Expression level도 RPKM으로 설정해줬습니다.
9) RNA-Seq Result

RNA-Seq 결과물은 Selected input sequence, References, Read Quality control, mapping statistics, fragment statistics, Distribution, Transcript length coverage 관련한 결과들을 확인할 수 있습니다.
분석이 제대로 되었는지 확인하기 위해 read quality control을 확인하였습니다.

Trimming을 하고 분석을 진행하였기에 분석 quality는 좋은 수치를 보였습니다.
그리고 결과물로 Gene expression, Transcript expression 결과에 관련하여서도 생성됩니다.
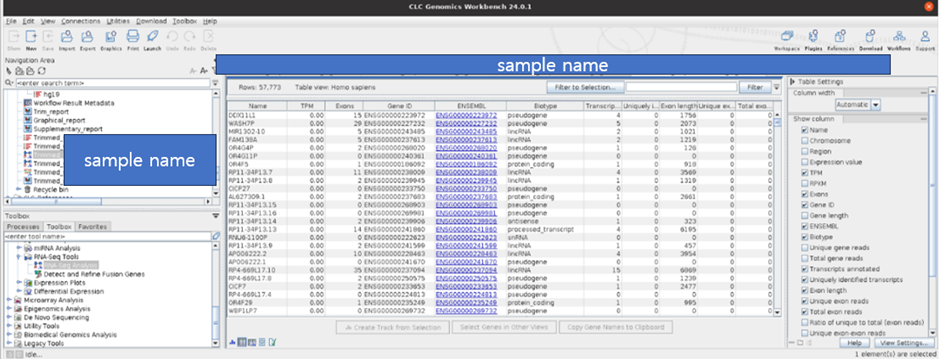
RNA Seq을 하는 목적 대부분은 Gene expression을 보기 위함으로 mRNA의 발현량을 측정할 수 있습니다. 이때 RPKM(Reads per kilobase per million reads) 또는 FPKM(Fragments Per Kilobase per Million reads) 등의 지표를 사용합니다.
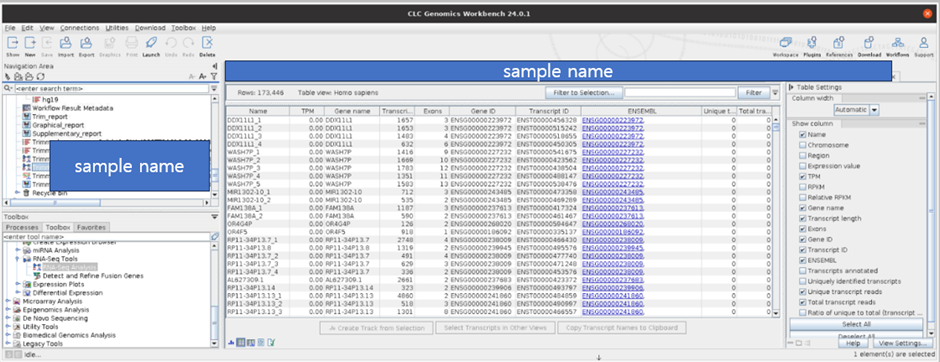
Transcription expression도 확인할 수 있습니다.
하지만 이번 분석의 목적은 RNA Fusion 확인이기 때문에 gene expression에 대해서는 자세하게 확인하지는 않았습니다.
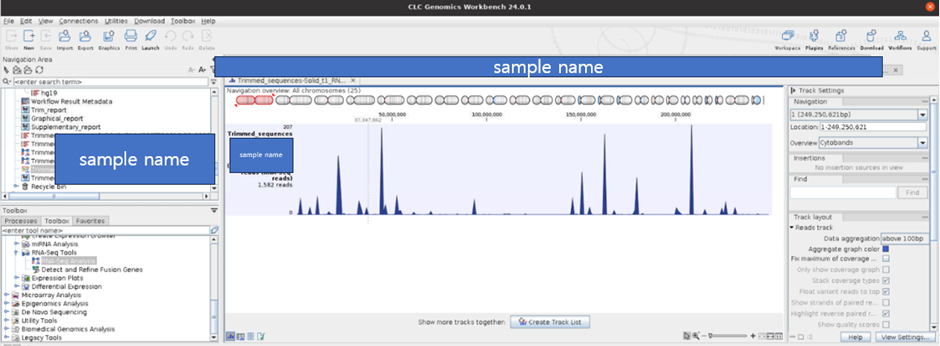
Mapping 된 결과물에 대한 전반적인 양상에 대해서도 visualization 해줍니다. 이를 통해 어떤 영역에 Depth가 많이 쌓였는지 볼 수 있습니다.
10) Fusion gene detection
RNA Fusion이란 유전자 재조합으로 인해 두 개의 유전자의 일부가 하이브리드 형태로 합쳐진 융합(Fusion) RNA를 생성하는 것을 의미합니다. 급성 백혈병, 폐암 등 일부 암종에서 유전자 재조합(rearrangement)이 종양발생에 중요한 역할을 하므로 이를 검출하는 것이 진단과 치료에 필수적입니다.
11) Fusion gene detection 방법
① 왼쪽 하단에 Toolbox에서 RNA-Seq tool인 Detection and Refine Fusion Genes를 클릭합니다. 이번에 확인할 데이터는 human의 solid tumor data입니다.
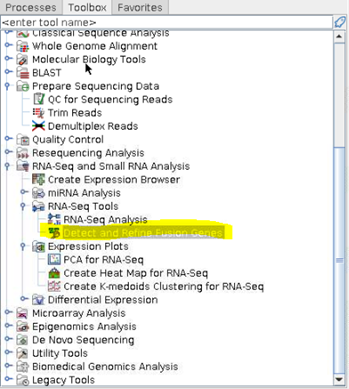
② 사용할 Solid tumor data를 선택합니다. 이때 RNA seq mapping data를 사용했습니다.
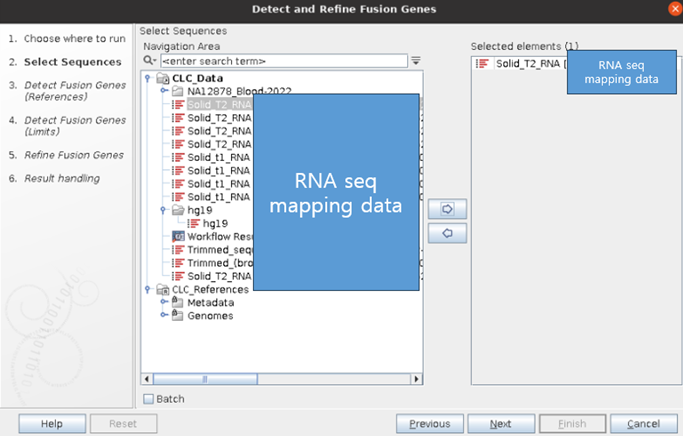
③ 아래와 같이 분석에 필요한 RNA mapping data, reference, mRNA track data, Gene track data를 함께 넣어줍니다.
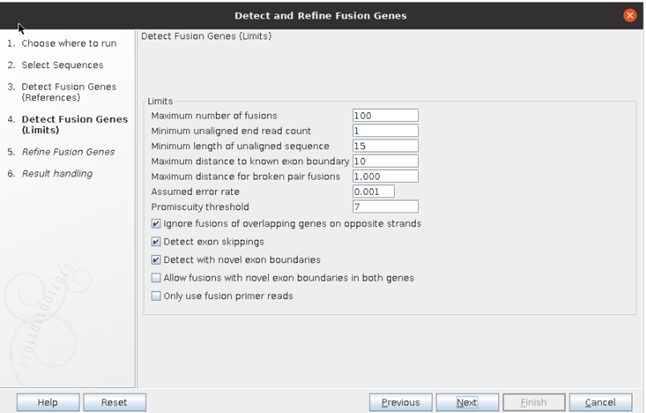
④ 분석 시, maximum number of fusion, read count 등 cut-off를 설정할 수 있는데, 아래와 같이 기본적으로 제공되는 cut-off를 설정하였습니다.
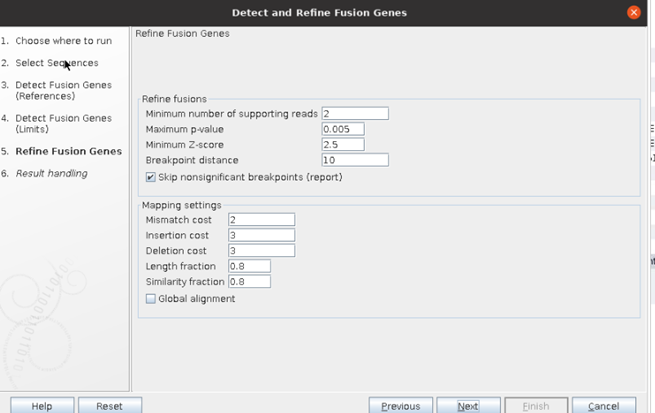
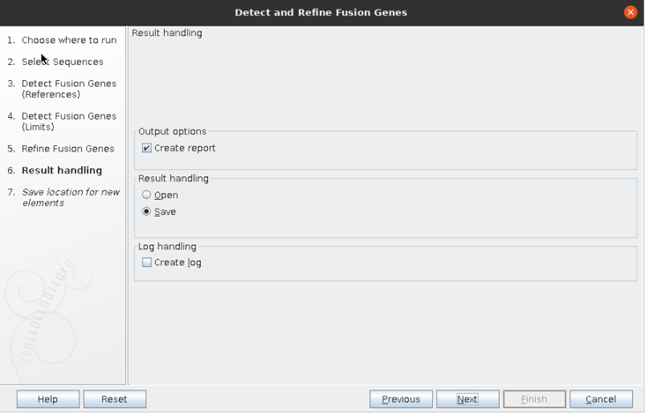
⑤ 마지막에 결과물을 따로 저장하고 싶으면, Save를 선택하고 결과물 확인만 하고 싶으면 Open을 선택하면 됩니다.
12) RNA Fusion Result
생성된 Fusion 결과물을 pdf로도 다운로드 받을 수 있습니다.

Fusion 결과 pdf에서 확인할 수 있는 내용은 Summary, Unaligned Ends, Fusion 정보들이며, 검출된 Fusion 들에 대한 자세한 정보를 확인할 수 있습니다.
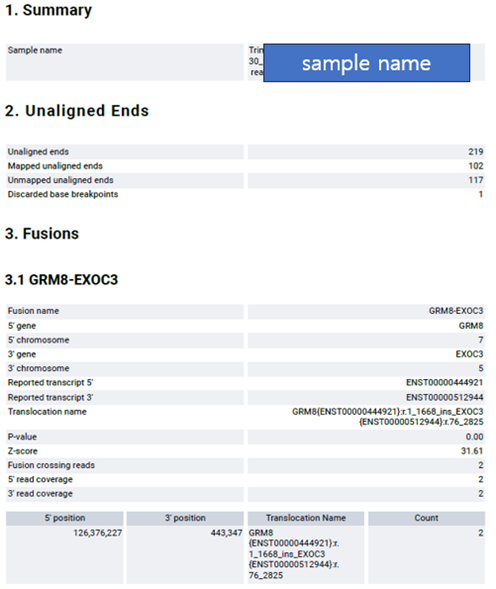
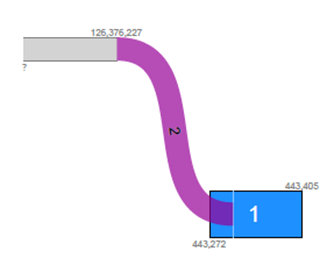
해당 샘플에서 GRM8-EXOC3가 fusion을 이루고 있음을 확인할 수 있었습니다. Fusion annotation 정보를 보면 5’ gene, 3’ gene 그리고 각 fusion의 exon 정보, Translocation name, read coverage 등을 확인할 수 있습니다.
해당 fusion의 read coverage는 2로 z-score도 31.61로 나온 것으로 확인됩니다. Read coverage가 너무 작아서 해당 fusion은 false call이 의심됩니다.
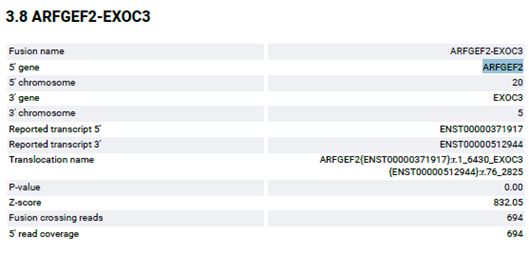
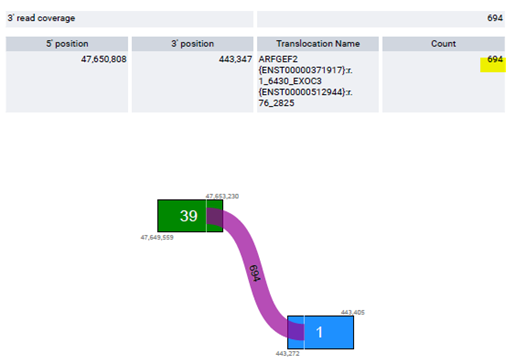
반면에 ARFGEF2-EXOC3 Fusion 결과의 경우 ARFGEF2 유전자의 20번 엑손과 EXOC3 유전자의 3번 엑손이 Fusion 되었으며, coverage depth가 694로 실제 BAM 파일로 확인하였을 때도 fusion이 확인됩니다.
4. 사용 후기
<장점>
평소에 다양한 생물정보 분석 Tool을 사용하고 있는데, tool 설치부터 파이프라인 세팅에 많은 시간이 소요됐었습니다. 인코렌탈을 이용하니 이러한 시간을 단축해서 더욱 연구에 집중할 수 있었어요. 또한 직접 코딩하지 않고 클릭만으로도 쉽게 분석하고 결과를 확인할 수 있다는 점, 그리고 분석 시간이 기존에 서버를 통해 사용하던 시간에 비해서 짧다는 것도 큰 장점이었습니다.
<단점>
아무래도 처음 사용할 때 데이터를 어떻게 업로드해야 하는지, 어떤 기능들이 어디에 존재하는지 익숙해지는 데까지 시간이 걸리는 것 같습니다. 사용자 친화적으로 구성된 서비스이지만 분석에 사용되는 format을 만들거나 생성되는 결과물이 어떤 결과를 하고 있는지 미리 파악하기가 어려운 것 같습니다.
분석 시간이 짧게 걸린다는 장점을 통하여 연구에 걸리는 시간을 단축해줄 것 같으며, RNA Fusion 결과도 한눈에 알아보기 쉽게 pdf로 시각화 및 정리되어 생성되어 샘플마다 비교하기 수월하였습니다. 그리고 Fusion의 경우에 어떤 exon끼리 연결되어있는지 확인하는 것이 중요한데 이 부분을 처음부터 시각화 해줘서 결과 해석하기에 유용하였습니다. 이렇게 인코렌탈을 이용하게 된다면 생물 정보 데이터 분석에 좀 더 도움이 될 것 같습니다.
'해당 포스팅은 업체로부터 제품과 원고료를 지원받아 실제 사용한 후기를 작성하였습니다.'
'Bioinformatics Issue' 카테고리의 다른 글
| NA12878 Download (FASTQ, VCF, BED) (0) | 2024.12.19 |
|---|---|
| [강연]유전자 가위 활용 질병 진단 기술 (0) | 2024.11.21 |
| [DB]What is JAX ClinicalKnowledgebase DB? (JAX CKB) (0) | 2024.04.03 |
| [BI]GATK HaplotyeCaller 사용해서 germline variant 확인하기 (1) | 2024.02.07 |
| HLA(Human Leukocyte Antigen)란 무엇인가? (0) | 2022.01.03 |